Molecular Evolution with lifesimulatoR
Introduction
One of the central questions in origin-of-life research is how non-living chemical systems could begin to exhibit life-like behaviour. Modern life relies on molecules that store information, make copies of themselves, mutate, and undergo selection. DNA and RNA perform these roles today, but early life-like systems may have been much simpler.
In lifesimulatoR, molecular evolution is represented
using symbolic sequences. A molecule may be represented by a sequence
such as "AUGCUA". The letters A,
U, G, and C are inspired by RNA
chemistry, but the model is conceptual rather than chemically
realistic.
The basic evolutionary workflow explored in this vignette is:
- Create a prebiotic molecular pool.
- Estimate molecular fitness.
- Replicate molecules.
- Introduce mutation.
- Apply selection.
- Repeat the process over many generations.
- Observe how the population changes.
Create a prebiotic molecular pool
Every evolutionary system requires a starting population. Before mutation, replication, or selection can occur, there must first be a collection of molecules capable of varying from one another.
In origin-of-life research, this starting collection is often called a prebiotic pool.
A prebiotic pool represents an early chemical environment containing
many different molecules. In reality these molecules could include amino
acids, nucleotides, peptides, lipids, and other organic compounds. In
lifesimulatoR, they are represented as symbolic
sequences.
pool <- create_prebiotic_pool(
n_molecules = 20,
alphabet = c("A", "U", "G", "C"),
min_length = 5,
max_length = 12,
seed = 123
)
head(pool)## [1] "GGUGUUUGACU" "AUGCAGGACA" "AGCUG" "AUGCUA" "GACGCUA"
## [6] "AAUGGCAGAGC"The output is a character vector where each element represents one symbolic molecule.
Why variation matters
Variation is essential because selection can only act when differences already exist.
If every molecule were identical, no molecule would have an advantage over any other.
Selection can only act on existing variation.
Understanding the parameters
-
n_molecules: number of molecules generated -
alphabet: symbols used to construct molecules -
min_length: minimum sequence length -
max_length: maximum sequence length -
seed: random seed for reproducibility
larger_pool <- create_prebiotic_pool(
n_molecules = 100,
alphabet = c("A", "U", "G", "C"),
min_length = 5,
max_length = 12,
seed = 123
)
length(larger_pool)## [1] 100Exploring the pool
length(pool)## [1] 20
nchar(pool)## [1] 11 10 5 6 7 11 10 5 5 8 12 8 12 7 7 11 10 10 12 6
data.frame(
molecule = pool,
length = nchar(pool)
)## molecule length
## 1 GGUGUUUGACU 11
## 2 AUGCAGGACA 10
## 3 AGCUG 5
## 4 AUGCUA 6
## 5 GACGCUA 7
## 6 AAUGGCAGAGC 11
## 7 AUAACCGAUA 10
## 8 GAUAG 5
## 9 GUCGC 5
## 10 UUGCUUGG 8
## 11 AUUAUCAAUGGA 12
## 12 UACUAGGC 8
## 13 UCGAUUCGUACG 12
## 14 GUUGAAC 7
## 15 UUUCUUU 7
## 16 CCUAUUUGCUG 11
## 17 GCAGGGAGGU 10
## 18 UGCGGUAUCC 10
## 19 AGGCCUCAGAGU 12
## 20 AGUCAG 6Calculate molecular fitness
Fitness is a simplified score representing how likely a molecule is to persist, replicate, or be selected.
In real chemistry, this would depend on factors such as:
- stability
- catalytic activity
- environmental conditions
- energy availability
example_sequence <- "AUGCUA"
molecule_fitness(example_sequence)## [1] 0.8876282Comparing fitness values
molecules <- c(
"AUGC",
"AAAAUUUU",
"GCGCGC",
"AUAUAUAUAUAU"
)
fitness <- molecule_fitness(molecules)
data.frame(
molecule = molecules,
fitness = fitness
)## molecule fitness
## 1 AUGC 0.6993290
## 2 AAAAUUUU 1.0687308
## 3 GCGCGC 0.8876282
## 4 AUAUAUAUAUAU 1.2500000Questions to consider:
- Should longer molecules have higher fitness?
- Should catalytic motifs increase fitness?
- How should fitness be represented in origin-of-life simulations?
Mutation: changing molecular sequences
Mutation introduces novelty into a molecular population.
Without mutation, populations may replicate, but they cannot easily explore new sequence space.
In origin-of-life models, mutation can represent:
- copying errors
- chemical modifications
- structural rearrangements
Mutation can be explored at two levels:
- Individual sequences
- Entire populations
Mutate one sequence
set.seed(2)
original <- "AUGCAUGCAUGC"
mutated <- mutate_sequence(
sequence = original,
alphabet = c("A", "U", "G", "C"),
mutation_rate = 0.2
)
data.frame(
original = original,
mutated = mutated
)## original mutated
## 1 AUGCAUGCAUGC UUGGAUACAUGCA mutation rate of 0.2 means each position has a
relatively high chance of being altered.
Low versus high mutation rate
set.seed(3)
low_mutation <- mutate_sequence(
sequence = "AUGCAUGCAUGC",
alphabet = c("A", "U", "G", "C"),
mutation_rate = 0.01
)
set.seed(3)
high_mutation <- mutate_sequence(
sequence = "AUGCAUGCAUGC",
alphabet = c("A", "U", "G", "C"),
mutation_rate = 0.40
)
data.frame(
mutation_rate = c(0.01, 0.40),
mutated_sequence = c(low_mutation, high_mutation)
)## mutation_rate mutated_sequence
## 1 0.01 AUGCAUGCAUGC
## 2 0.40 GUCCAUCCAUGCA low mutation rate usually preserves the original sequence.
A high mutation rate introduces more variation, but excessive mutation may disrupt useful molecular patterns.
Variation is necessary for evolution, but too much variation can prevent useful information from being preserved.
Mutate a population
set.seed(4)
molecules <- c("AUGC", "UUUU", "GCGC", "AAAA")
mutated_population <- mutate_population(
molecules = molecules,
mutation_rate = 0.2
)
data.frame(
before = molecules,
after = mutated_population
)## before after
## 1 AUGC AGGC
## 2 UUUU UUUU
## 3 GCGC UCGU
## 4 AAAA AAAASome molecules remain unchanged, while others accumulate mutations.
Replication and selection
Replication allows successful molecules to become more common.
Selection means that molecules with higher fitness have a greater chance of contributing to future generations.
molecules <- c(
"AUGC",
"AAAAUUUU",
"GCGCGC",
"AUAUAUAUAUAU"
)
next_generation <- replicate_molecules(
molecules = molecules,
n_molecules = 20,
selection_strength = 1
)
next_generation## [1] "GCGCGC" "GCGCGC" "AUGC" "AAAAUUUU" "AAAAUUUU"
## [6] "GCGCGC" "AUGC" "AAAAUUUU" "AUGC" "AAAAUUUU"
## [11] "AAAAUUUU" "AAAAUUUU" "AUAUAUAUAUAU" "AUGC" "GCGCGC"
## [16] "AAAAUUUU" "AUGC" "AAAAUUUU" "GCGCGC" "AAAAUUUU"Selection strength
The parameter selection_strength controls how strongly
fitness influences replication.
-
0= neutral drift - low values = weak selection
- high values = strong selection
Compare neutral drift and selection
set.seed(1)
neutral <- replicate_molecules(
molecules = molecules,
n_molecules = 100,
selection_strength = 0
)
set.seed(1)
selected <- replicate_molecules(
molecules = molecules,
n_molecules = 100,
selection_strength = 2
)
table(neutral)## neutral
## AAAAUUUU AUAUAUAUAUAU AUGC GCGCGC
## 20 21 27 32
table(selected)## selected
## AAAAUUUU AUAUAUAUAUAU AUGC GCGCGC
## 29 37 11 23As selection strength increases, fitter molecules become more common.
Evolve one generation
The function evolve_generation() combines:
- fitness
- replication
- mutation
- selection
into a single evolutionary step.
next_generation <- evolve_generation(
molecules = pool,
mutation_rate = 0.02,
selection_strength = 1
)
head(next_generation)## [1] "GGUGUUUGACU" "AUAACCGAUA" "AAUGGCAGAGC" "AGUCAG" "UUGCUUGG"
## [6] "CCUAUUUGCUG"One generation illustrates the mechanism. Many generations reveal longer-term trends.
Simulate abiogenesis-like molecular evolution
The main simulation function is
simulate_abiogenesis().
It starts with a random molecular pool and repeatedly applies:
- replication
- mutation
- selection
over many generations.
sim <- simulate_abiogenesis(
n_molecules = 100,
generations = 200,
mutation_rate = 0.01,
selection_strength = 1,
seed = 10
)
head(sim)## # A tibble: 6 × 6
## generation n_molecules mean_length mean_fitness diversity max_fitness
## <int> <int> <dbl> <dbl> <int> <dbl>
## 1 0 100 12.6 1.02 100 1.25
## 2 1 100 12.4 1.08 69 1.25
## 3 2 100 12.7 1.08 58 1.25
## 4 3 100 12.7 1.11 55 1.25
## 5 4 100 13.0 1.12 53 1.25
## 6 5 100 13.0 1.13 47 1.25
tail(sim)## # A tibble: 6 × 6
## generation n_molecules mean_length mean_fitness diversity max_fitness
## <int> <int> <dbl> <dbl> <int> <dbl>
## 1 195 100 12 1.25 35 1.25
## 2 196 100 12 1.25 37 1.25
## 3 197 100 12 1.25 37 1.25
## 4 198 100 12 1.25 39 1.25
## 5 199 100 12 1.25 42 1.25
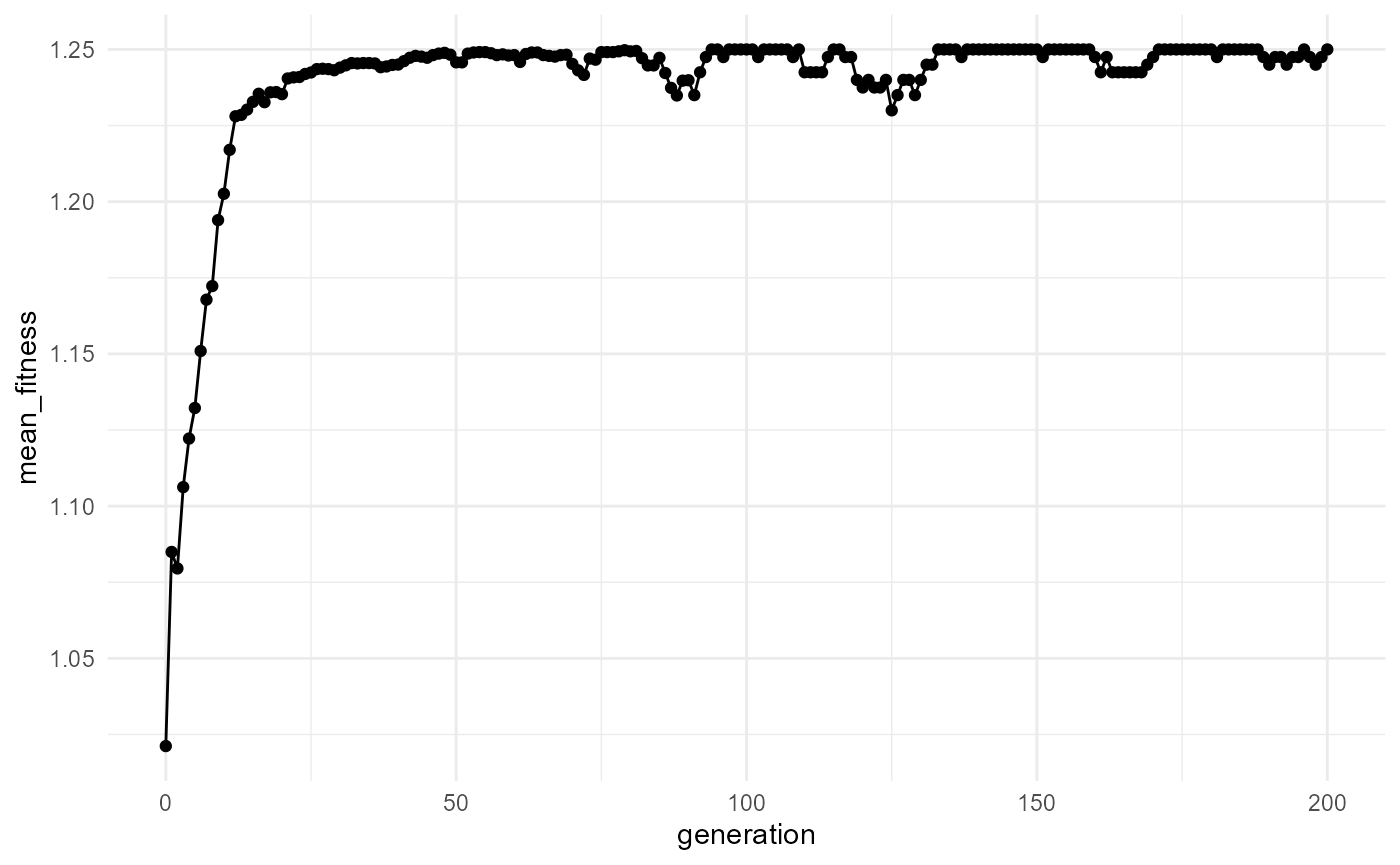
## 6 200 100 12 1.25 39 1.25The output is a tibble summarizing population-level changes through time.
Visualize results
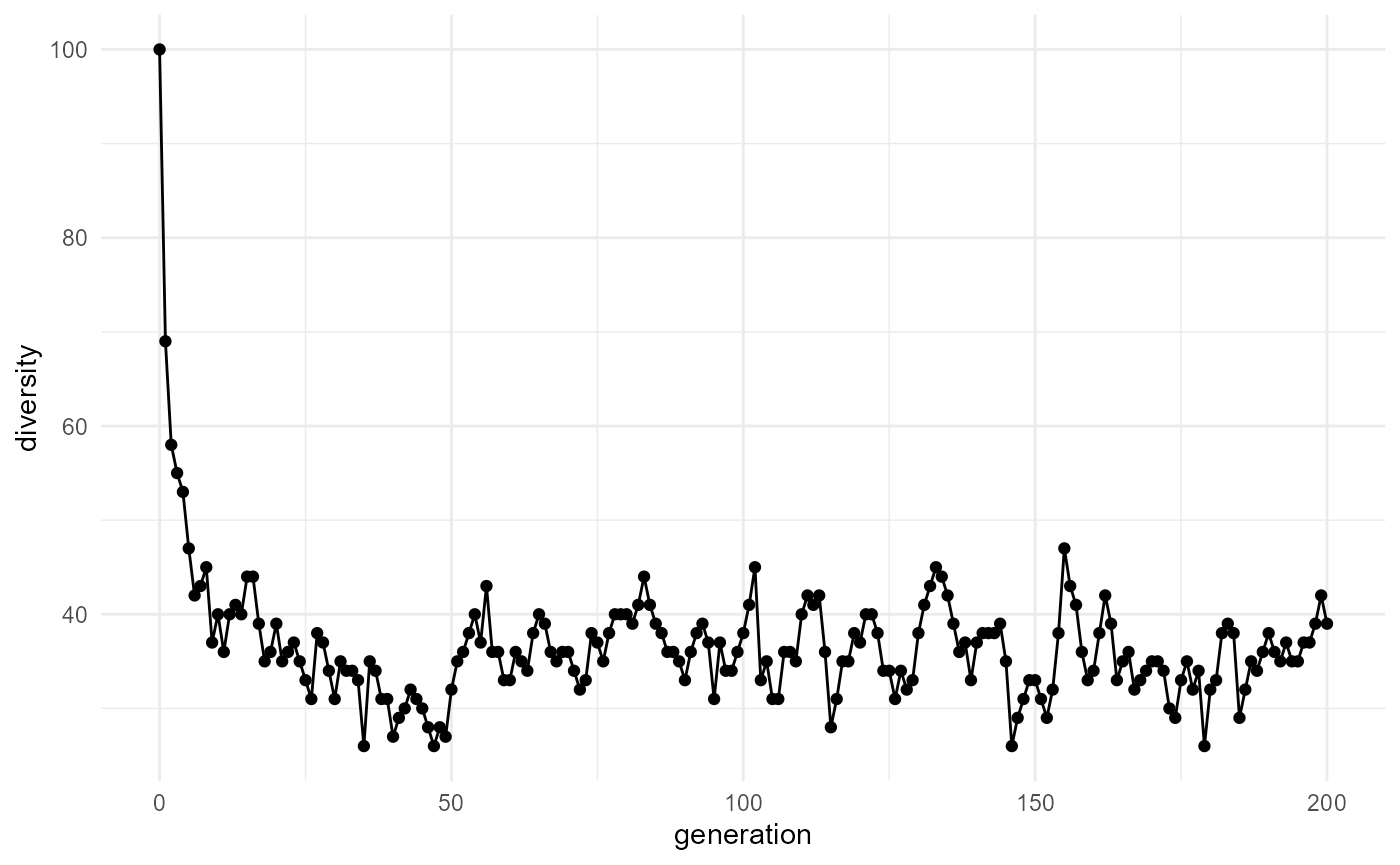
Plot diversity
plot_simulation(
sim,
x = "generation",
y = "diversity"
)
Plots can help answer questions such as:
- Does mean fitness increase?
- Does diversity increase or decrease?
- Does the population stabilize?
- Does selection lead to dominance by certain molecule types?
Parameter experiments
Experiment 1: mutation rate
low_mutation <- simulate_abiogenesis(
n_molecules = 100,
generations = 100,
mutation_rate = 0.005,
selection_strength = 1,
seed = 123
)
high_mutation <- simulate_abiogenesis(
n_molecules = 100,
generations = 100,
mutation_rate = 0.10,
selection_strength = 1,
seed = 123
)Compare the results.
Questions:
- Which simulation produces more diversity?
- Which preserves information more effectively?
Experiment 2: selection strength
weak_selection <- simulate_abiogenesis(
n_molecules = 100,
generations = 100,
mutation_rate = 0.02,
selection_strength = 0.2,
seed = 123
)
strong_selection <- simulate_abiogenesis(
n_molecules = 100,
generations = 100,
mutation_rate = 0.02,
selection_strength = 3,
seed = 123
)Questions:
- Does stronger selection increase fitness?
- Does stronger selection reduce diversity?
Interpretation
This tutorial demonstrates a simplified model of molecular evolution.
Key concepts include:
- Variation
- Fitness
- Replication
- Mutation
- Selection
- Population-level change
The model is intentionally simple. It does not simulate:
- real chemistry
- RNA folding
- thermodynamics
- energy flow
- detailed reaction kinetics
Instead, it provides an educational framework for exploring how life-like evolutionary dynamics may emerge.
Suggested exercises
- Run simulations with mutation rates of 0, 0.01, 0.05, and 0.20.
- Compare weak and strong selection.
- Change the alphabet used to generate molecules.
- Compare short and long molecule populations.
- Modify the fitness function.
- Compare neutral drift and selection.
- Discuss what would be needed to make the model chemically realistic.
- Compare molecular evolution with protocell models.
- Compare molecular evolution with autocatalytic network models.
- Debate whether replication-first, metabolism-first, or compartment-first models provide the best explanation for the origin of life.